Understanding Cleanroom Classifications
Over the past few years there has been an increasing trend to change from previous classification systems used to the ISO classification systems in ISO 14644-1. However, many companies have continued to use the traditional Class 100, 10,000, 100,000 room classification system from Federal Standard 209-e. In Europe, the GMPs as stated in Annex 1 utilize another system—Grades A through D.
Many global companies choose to use this classification system. All of these systems are acceptable for use. However, we have also tended to link the systems together, e.g., ISO 5/Class 100/Grade A. This type of linkage is seen in the FDA’s Guidance for Aseptic Processing (2004). If you are manufacturing an aseptic product and use this linked classification system it is not likely to be an issue. However, if you are not manufacturing an aseptically processed product, choosing to link the classification systems together may lead to other consequences.
Federal Standard 209e
This document was written for use by federal agencies of the United States. The scope of this document is defined as:
“This document establishes standard classes, and provides for alternative classes, of air cleanliness for cleanrooms and clean zones based on specified concentrations of airborne particles. It prescribes methods for verifying air cleanliness and requires that a plan be established for monitoring air cleanliness. It also provides a method for determining and describing concentrations (U descriptors) of ultrafine particles”
Equally important are the limitations identified in this document, including:
“The requirements of this document do not apply to equipment or supplies for use within cleanrooms or clean zones. Except for size classification and population, this document is not intended to characterize the physical, chemical, radiological, or viable nature of airborne particles. No universal relationship has been established between the concentration of airborne particles and the concentration of viable airborne particles. In addition to the need for a clean air supply that is monitored for total particulate contamination and that meets established limits, special requirements are necessary for monitoring and controlling other forms of contamination.”
Based upon these limitations, no requirements existed for the viable microorganisms allowed to be present in support of these classification systems. This system utilized classifications Class 10/Class 100/Class 10,000 and Class 100,000. It also included references to an “M” system classification.
This document was subsequently retired and replaced by ISO 14644-1.
ISO 14644-1 classification system
ISO 14644-1, Cleanrooms and associated controlled environments-Part 1: Certification of Air Cleanliness document, was formally issued in 1999. This document establishes the certification requirements for air cleanliness areas. This document has replaced the old Federal Standard 209-e (Class 100, 10,000 and 100,000 designations). Within this document the various classification systems are based upon the requirements for counts associated with non-viable particulates. The limits stated in this document are depicted in Table 1.
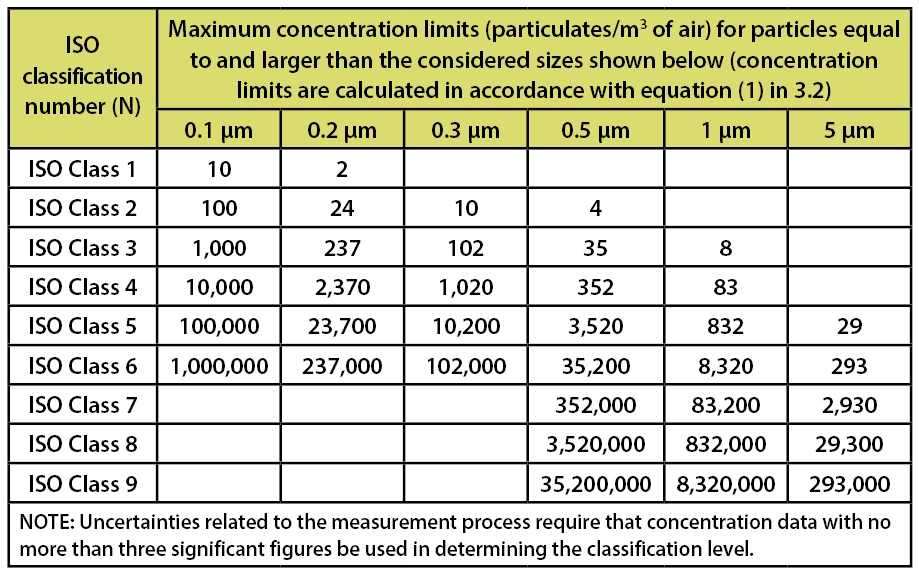
Table 1: Selected airborne particulate cleanliness classes for cleanrooms and clean zones. Classification Parameters from ISO 14644-1.
This document does not include specific requirements for sterile or non-sterile product, nor does it include requirements for any parameter excluding non-viable particulates. As such, unlike the FDA’s Aseptic Processing Guidance and the EU’s GMPs Annex 1, there are no specified limits for viable microorganisms present.
FDA’s Aseptic Processing Guidance
In the FDA’s Guidance for Industry—Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice (2004)—which is limited in scope to the manufacture of medicinal products using aseptic processing—there is a similar chart which includes the requirements for both viable and non-viable microbial counts as part of the classification system. This classification system is depicted in Table 2.
Footnote “b” of this table indicates that ISO 5 particle concentration is equal to Class 100 and approximately equal to EU Grade A. However the microbiological limits in this document are only applicable to aseptic processing. This statement has led many to arbitrarily equate ISO 5, Class 100, and Grade A routinely. This statement of “equality” is frequently shown in published literature and industry documents.
The European Union’s GMPs – Annex 1
For the European Union, the Drug GMPs are part of EudraLex, The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. This document is supplemented by Annex 1 Manufacture of Sterile Medicinal Products (corrected version). Annex 1 applies to drug products manufactured by aseptic processing and/or terminal sterilization. Note: Annex 1 section 5 states: “For classification purposes EN/ISO 14644-1 methodology defines both the minimum number of sample locations and the sample size based on the class limit of the largest considered particle size and the method of evaluation of the data collected.” This document includes a table of limits for microbial contamination. This data is included in Table 3
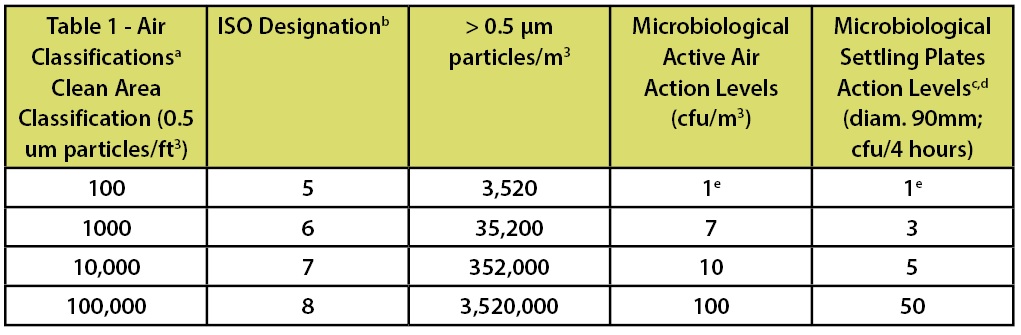
Table 2: Classification Parameters from the FDA’s Aseptic Processing Guidance
While the tables indicate that these are recommended limits, there is a clear expectation from European investigators that companies will meet these limits on a consistent basis if they are going to claim a specific classification for a room.
Additionally, Annex 1 section 9 indicates:
“For Grade A zones, particle monitoring should be undertaken for the full duration of critical processing, including equipment assembly, except where justified by contaminants in the process that would damage the particle counter or present a hazard, e.g. live organisms and radiological hazards. In such cases monitoring during routine equipment set up operations should be undertaken prior to exposure to the risk. Monitoring during simulated operations should also be performed. The Grade A zone should be monitored at such a frequency and with suitable sample size that all interventions, transient events and any system deterioration would be captured and alarms triggered if alert limits are exceeded. It is accepted that it may not always be possible to demonstrate low levels of ≥5.0 μm particles at the point of fill when filling is in progress, due to the generation of particles or droplets from the product itself.”
In addition to requirements for microbial contamination, Annex 1 contains a table to specify the requirements for particulate contamination. Table 4 summarizes these requirements.
Annex 2 of the European Union’s GMPs is entitled Manufacture of Biological Active Substances and Medicinal Products for Human Use. Within this document, item 6 is relevant to the use of controlled environments in the manufacturing process:
6. “Manufacturing and storage facilities, processes and environmental classifications should be designed to prevent the extraneous contamination of products. Prevention of contamination is more appropriate than detection and removal, although contamination is likely to become evident during processes such as fermentation and cell culture. Where processes are not closed and there is therefore exposure of the product to the immediate room environment (e.g., during additions of supplements, media, buffers, gases, manipulations during the manufacture of ATMPs) control measures should be put in place, including engineering and environmental controls on the basis of QRM principles. These QRM principles should take into account the principles and guidance from the appropriate sections of Annex 1 to EudraLex, Volume 4, when selecting environmental classification cascades and associated controls.
ICH Q7A
ICH Q7 is the Good Manufacturing Practice for Active Pharmaceutical Ingredients issued by the International Conference on Harmonisation Regulations. This is a recognized document for both the United States and Europe. The scope of this document states: “This Guide applies to the manufacture of APIs for use in human drug (medicinal products). It applies to the manufacture of sterile APIs only up to the point immediately prior to the APIs being rendered sterile. The sterilization and aseptic processing of sterile APIs are not covered by this guidance…” As such, it relates to the manufacture of non-sterile APIs and sterile APIs during the non-sterile stages.
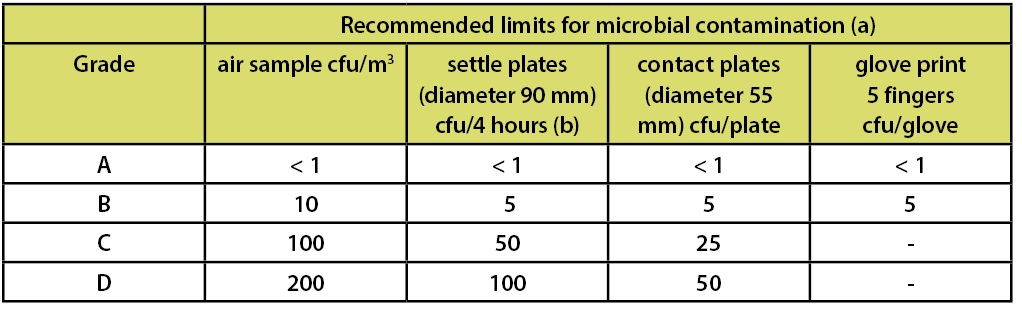
Table 3: EU GMPs Annex 1 Recommended Limits for Microbial Contamination
Section 8.5 of ICH Q7 is entitled Contamination Control. The requirements as stated in this document are:
“8.50 Residual materials can be carried over into successive batches of the same intermediate or API if there is adequate control. Examples include residue adhering to the wall of a micronizer, residual layer of damp crystals remaining in a centrifuge bowl after discharge, and incomplete discharge of fluids or crystals from a processing vessel upon transfer of the material to the next step in the process. Such carryover should not result in the carryover of degradants or microbial contamination that may adversely alter the established API impurity profile.
“8.51 Production operations should be conducted in a manner that will prevent contamination of intermediates or APIs by other materials.”
“8.52 Precautions to avoid contamination should be taken when APIs are handled after purification.”
This section has no requirements for the use of classified areas nor does it have specific requirements for microbial monitoring of the processes.
Section 18 of this document provides specific guidance for APIs manufactured by cell culture/fermentation.
Section 18.15 states:
“Appropriate and environmental controls should be used to minimize the risk of contamination. The acceptance criteria for quality of the environment and the frequency of monitoring should depend on the step in production, and the production conditions (open, closed or contained systems).”
The following requirements are applicable to the cell culture/fermentation process (relative to air classification and/or environmental monitoring and harvesting, isolation and purification steps):
“18.30 Where aseptic addition of cell substrates, media, buffers, and gases is needed, closed or contained systems should be used where possible. If the inoculation of the initial vessel or subsequent transfers or additions (media, buffers) are performed in open vessels, there should be controls and procedures in place to minimize the risk of contamination.”
“18.31 Where the quality of the API can be affected by microbial contamination, manipulations using open vessels should be performed in a biosafety cabinet or similarly controlled environment.”
“18.40 Harvesting steps, either to remove cells or cellular components or to collect cellular components after disruption should be performed in equipment and areas designed to minimize the risk of contamination.”
“18.43 If open systems are used, purification should be performed under environmental conditions appropriate for the preservation of product quality.”
Reviewing all of the ICH Q7 requirements, there are no designated systems for classification of biosafety cabinets or laminar airflow hoods. Additionally there are no specified requirements for microbiological monitoring of these areas other than they should be appropriate to minimize risk of contamination and provide appropriate conditions for preserving product quality.
Aseptic processes
When using these documents, the “linked” form of Class 100/ISO 5/Grade A can be easily used. Although there are some small differences in the documents, these differences can be easily resolved, e.g., microbiological limits of 1 cfu in the aseptic guidance and <1cfu in the European GMPs. The difference in this case being whether you can average results to determine whether the limits are met for each parameter. Usually the tightest limit is used.
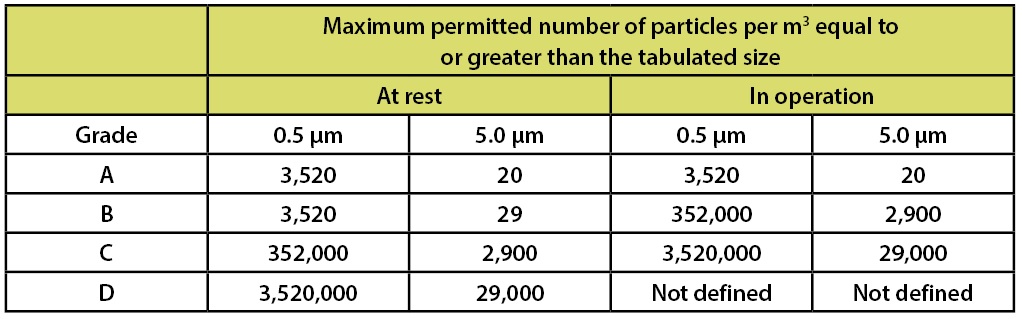
Table 4: EU GMPs Annex 1 Recommended Limits for Particulate Contamination
The problem
So many companies have seen the linked classifications, e.g. Class 100/ISO 5/Grade A, that they routinely use this classification system in their facility regardless of the type of manufacturing process. If, for example, you are manufacturing a non-sterile bulk drug or a terminally sterilized drug and you utilize this classification link, you are choosing to utilize the microbiological limits for Grade A, which were designed for the production of sterile medicinal products. Additionally, non-sterile manufacturers rarely have in place the other requirements of a Grade A facility including things like: changing rooms, cascading room classification, gowning requirements, and the microbiological control limits and frequencies. It isn’t even likely that the process required the level of control specified for Grade A.
This scenario changes somewhat if the non-sterile bulk is also a biologic product. In Annex 2, it indicates that one needs to take into account the requirements for classification and cascading room classifications specified in Annex 1. There are provisions to utilize quality risk management procedures when considering this requirement. For example, you may have a risk analysis and control program that shows the microbiological control procedures may not be required for your process at the levels stated for the specified room grade.
For companies that manufacture products for both the United States and Europe, should you choose to utilize a classification system other than the Grades A through D it is crucial that you have a document that explains the relationship of your classification system to the European classification system.
It is very important that you understand the implications of the classification system you use and to use them wisely.
References:
• EU (2008) EudraLex The Rules Governing Medicinal Products in the European Union Volume 4
• EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use.
• Annex 1 Manufacture of Sterile Medicinal Products (corrected version). European Commission. Brussels. 25 November 2008 (revised).
• EU (2012) EudraLex The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Human and Veterinary Use. Annex 2 Manufacturer of Biological Active Substances and Medicinal Products for Human Use. European Commission. Brussels. Deadline for coming into compliance 31 January 2013. (SANCO/AM/sl/ddg1.d.6(2012)860362)
• FED-STD-209e (1992) Federal Standard 209-e Airborned Particulate Cleanliness Classes in Cleanrooms and Clean Zones. This standard is approved by the Commissioner, Federal Supply Service, General Services Administration, for the use of all Federal Agencies.
• FDA (2004) Guidance for Industry, Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice. U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Office of Regulatory Affairs (ORA) Pharmaceutical CGMPs
• ISO (1999) International Standard 14644-1: Cleanrooms and associated controlled environments-Part 1: Certification of Air Cleanliness. International Organisation for Standardisation. Switzerland.
• ISO (2003) International Standard ISO 14698-2:2003 Cleanrooms and associated controlled environments — Biocontamination control —Part 2: Evaluation and interpretation of biocontamination data
• FDA (2001) ICH Q7A Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients
Jeanne Moldenhauer is the Vice President of Excellent Pharma Consulting. She has over 25 years of experience in the pharmaceutical, biotech, and device industries. She is very active in PDA, has authored many books and articles, and can be reached at info@excellpharma.com.
This article appeared in the March 2014 issue of Controlled Environments.














